We previously identified LMTK3 as a regulator of ERα with prognostic & predictive significance for breast cancer (BC) patients’ survival, also possessing roles in intrinsic & acquired endocrine resistance.
We also reported the association of LMTK3 in promoting invasion & migration in BC and its scaffolding function in transcriptional regulation.
Recently, we solved the crystal structure of LMTK3 kinase domain, determined its consensus motif and reported that LMTK3 is a HSP90 client protein. We also identified a potent LMTK3 inhibitor that confers cytotoxic & antitumor effects.
Taken together, we believe that further deciphering the pathways that LMTK3 is implicated in and the cross-talk with other proteins will reveal fundamental insights into its role in endocrine resistance and metastasis in breast cancer (and potentially other types of cancer and diseases) and derive new druggable targets.
In aggregate, the development of oral LMTK3 inhibitors may have the potential for broad clinical use, either as monotherapy or as a combinational therapy.
- PAPER: Identification of LMTK3 as a new therapeutic target in breast cancer (Nature Medicine, 2011; BCRT, 2011)
-
Over two-thirds of breast tumours express the Estrogen Receptor-alpha (ERα) and patients with ERα-positive disease respond to anti-estrogens (tamoxifen-(Tam)), estrogen withdrawal (aromatase inhibitors) or ERα downregulation (fulvestrant). However, resistance frequently occurs with tumours recurring as metastatic. Moreover, Tam-resistant ERα-positive cells exhibit a transition towards a more aggressive phenotype displaying augmented motility and invasiveness. Accumulating evidence suggests that ERα extra-nuclear signalling (cross-talk with kinases) can promote cell migration and metastasis.
By performing a whole human kinome siRNA screening, we identified Lemur-Tyrosine-Kinase-3 (LMTK3) as a regulator of ERα with prognostic and predictive significance for breast cancer patients’ survival, also possessing roles in intrinsic and acquired endocrine resistance (Fig. 1). Interestingly, LMTK3 has undergone Darwinian positive selection compared with its chimpanzee ortholog, a noteworthy result given the unique susceptibility of humans to epithelial neoplasms, including breast cancer .
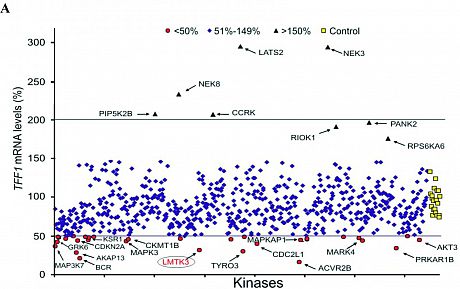
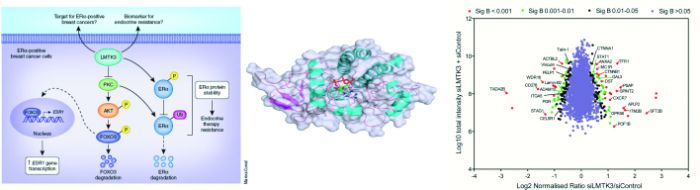
Fig. 1 Identification of LMTK3 as an ERα regulator and its association with clinical outcome in breast cancer .
A) High-throughput siRNA screen identifying kinases modulating ERα transcriptional activity, using the mRNA expression
levels of TFF1 as read-out. Kaplan-Meier plots showing the association between LMTK3 expression and (B) disease-free
survival (P = 0.012) and (C) response to endocrine therapy (P = 0.039).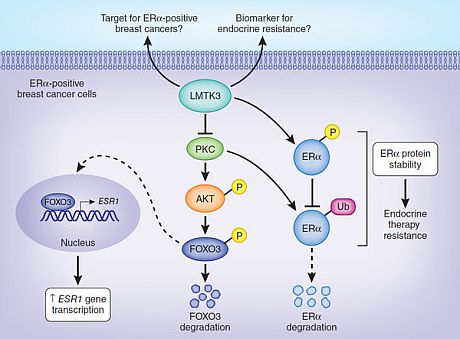 MechanisticallyLMTK3 enhances transcriptional activation of ESR1 by inhibiting PKC, whose activity results in phosphorylation of AKT, which in turn phosphorylates FOXO3, targeting it for degradation. LMTK3 also directly phosphorylates ERα protein, which has been shown to contribute to resistance to endocrine therapies such as tamoxifen. LMTK3 protects ERα from ubiquitin (Ub)-mediated proteasomal degradation directly through phosphorylation and indirectly through inactivation of PKC (Fig. 2).
MechanisticallyLMTK3 enhances transcriptional activation of ESR1 by inhibiting PKC, whose activity results in phosphorylation of AKT, which in turn phosphorylates FOXO3, targeting it for degradation. LMTK3 also directly phosphorylates ERα protein, which has been shown to contribute to resistance to endocrine therapies such as tamoxifen. LMTK3 protects ERα from ubiquitin (Ub)-mediated proteasomal degradation directly through phosphorylation and indirectly through inactivation of PKC (Fig. 2). Fig. 2 LMTK3 positively regulates ESR1 gene transcription,
protein levels and phosphorylation, promoting resistance
to endocrine therapies. - PAPER: LMTK3 promotes invasion, migration and actin cytoskeleton remodeling (Science Signaling; 2014)
-
Cell migration and invasion are the early steps of the metastatic process controlled by cellular signals that stimulate changes in cell adhesion and cytoskeleton formation.
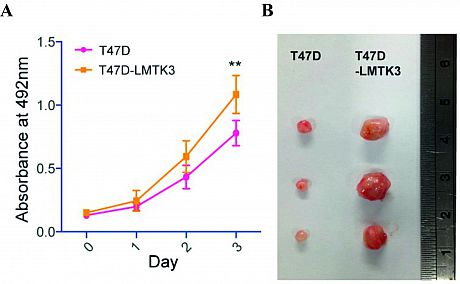
We have demonstrated that LMTK3 overexpression promotes breast cancer cell proliferation in culture and accelerates tumor growth in vivo in a breast tumor xenograft model after injecting T47D or T47D-LMTK3 over-expressing cells into the mammary fat pads of female Balb/C nude mice (Fig. 1).
Fig 1.LMTK3 promotes breast cancer growth in vitro and in vivo.
(A) Effect of overexpression of LMTK3 on the proliferation in T47D
cells compared to the parental cell lines. (B) Quantification of fold
difference in tumor size between T47D and T47D-LMTK3 xenografts
at weeks 2 & 6. Representative images of tumors at 6weeks are shown.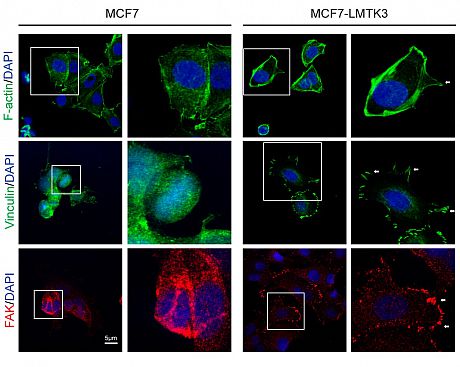 Adhesion to the ECM results in the generation of contractile forces and dynamic actin polymerization and re-organization, which are some of the major remodeling changes that occur in the cell during migration and invasion.
Adhesion to the ECM results in the generation of contractile forces and dynamic actin polymerization and re-organization, which are some of the major remodeling changes that occur in the cell during migration and invasion. We found that overexpression of LMTK3 increased actin-rich protrusion and focal adhesion formations on the edge of migrating cells in both 2D and 3D cultures (Fig 2).
Fig 2. LMTK3 regulates actin cytoskeleton. Confocal
microscopy analysis of F-actin protrusions, viniculin
localization, and FAK abundance in MCF7 & MCF7-LMTK3
cells.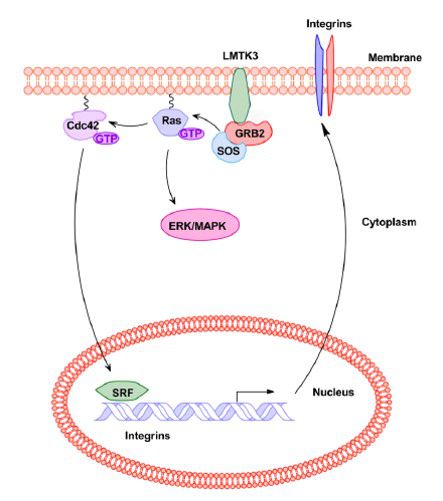 In our proposed model, LMTK3 promotes invasion in breast cancer through GRB2-mediated induction of integrin b1.
In our proposed model, LMTK3 promotes invasion in breast cancer through GRB2-mediated induction of integrin b1.More precisely,GRB2 silencing in cell lines overexpressingLMTK3 suppressed integrin-encoding gene expression, as well as cell adhesion and invasion. SRF inhibition effectively inhibited LMTK3-induced migration but did not suppress LMTK3-induced proliferation, implying that LMTK3 regulates cell proliferation through GRB2 through the RAS-ERK/MAPK pathway and cell motility through the CDC42-SRF pathway, which results in enhanced ITGB1 and ITGA5 expression (Fig. 3).
Fig 3 . Model of LMTK3 signalling in promoting breast cancer invasion. - PAPER: LMTK3 functions as a transcriptional co-repressor of tumor suppressor-like genes (Cell Reports; 2015)
-
Several receptor tyrosine kinases (RTKs) have been reported to localize in the nucleus, where they can regulate gene expression (most likely trans-activation) through binding to euchromatin. According to these reports, nuclear RTKs are present in the nucleoplasm instead of the nuclear lamina. In most cells, at least one class of heterochromatin is positioned at the nuclear lamina, resulting in gene repression. Previous studies also propose that heterochromatin relocation to the nuclear lamina might occur via active tethering mediated by discrete molecular complexes.
 We investigated the function of nuclear LMTK3 through mapping genome-wide chromatin interaction sites of LMTK3 in breast cancer.
We investigated the function of nuclear LMTK3 through mapping genome-wide chromatin interaction sites of LMTK3 in breast cancer.We observed that LMTK3 binding in MCF7 (ERα-positive) and MDA-MB-231 (ERα-negative) have a high similarity with a high correlation.
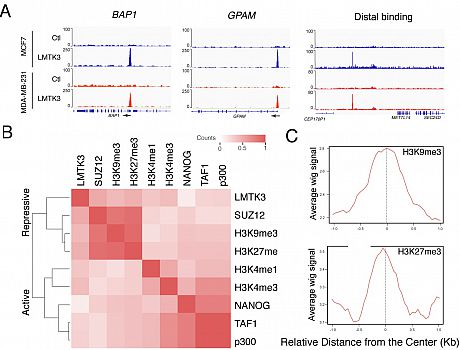
Interestingly, we found that LMTK3 binds chromatin at both repressive and active promoters, suggesting that there is a different binding profile of LMTK3 compared with the ones of other knownRTKs (Fig. 1).
Fig 1 . Identification of Genome-wide LMTK3 Binding Sequences with ChIP-Seq. (A) Binding of LMTK3 at the promoter of
BAP1, GPAM and the distal interval in MCF7 and MDA-MB-231 cells. (B) Clustering of genome-wide binding datasets with LMTK3.
(C) H3K9me3 & H3K27me3 enrichment around LMTK3 peaks in MCF7 cells.We further identified KAP1 (TRIM28) as a binding partner of LMTK3. The LMTK3/KAP1 interaction is stabilized by PP1a, which suppresses KAP1 phosphorylation specifically at LMTK3-associated chromatin regions, inducing chromatin condensation and resulting in transcriptional repression of LMTK3- bound tumor suppressor-like genes. Furthermore, LMTK3 functions at distal regions in tethering the chromatin to the nuclear periphery, resulting in H3K9me3 modification and gene silencing. In summary, we propose a model where a scaffolding function of nuclear LMTK3 promotes cancer progression through chromatin remodeling (Fig. 2).
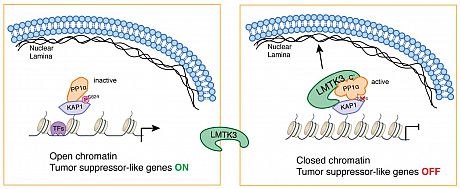
Fig 2. Graphical Summary of Chromatin Remodeling and Transcriptional Co-repressor Behavior of LMTK3.
A schematic illustration of mechanism of chromatin remodeling mediated by nuclear LMTK3. LMTK3 binds
PP1a through its C-terminal domains and interacts with KAP1 and dephosphorylates KAP1 at Ser824, which
results in chromatin condensation. Meanwhile, a part of the LMTK3 disordered domain tethers the whole
heterochromatin complex to the nuclear lamina through interacting with Lamin A. These result in the
transcriptional repression of LMTK3-bound tumor suppressor-like genes. - PAPER: Involvement of LMTK3 in chemo-resistance in breast cancer (Oncogene, 2018)
-
Adjuvant and neo-adjuvant chemotherapy still represent the backbone of systemic treatment for many cancers at both the early and advanced stages of the disease. DNA-damaging agents are used in cancer chemotherapy, as DNA integrity is crucial for proper cellular function and proliferation. One of these drugs is doxorubicin, an anthracycline antibiotic, which intercalates into DNA, hindering topoisomerase II progression and leading to cytotoxicity mostly by inhibition of DNA replication and generation of double-strand breaks (DSBs). However, even though doxorubicin is a widely used drug in chemotherapy regimen for breast cancer patients, resistance (innate and/or acquired) is often observed [22]. Therefore, cellular mechanisms that relate to doxorubicin-related resistance need deciphering.
In this study, we implicate LMTK3 in the protection of breast cancer cells from DNA DSBs as induced by doxorubicin. Using 2D monolayers as well as 3D-spheroids cultures we show that breast cancer cells stably overexpressing LMTK3 are more resistant to doxorubicin treatment when compared to the parental ones. Importantly, we underpin these results by using a breast cancer xenograftmouse model revealing that increased LMTK3 levels decrease the antitumor activity of doxorubicin (Fig 1).
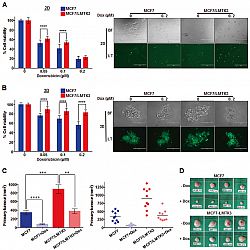 Fig 1.Over-expression of LMTK3 impedes the effectiveness of doxorubicin in vitro and in vivo. Following treatment with different concentrations of doxorubicin (0.05, 0.1, and 0.2 μM) for 72 h, the percentage (%) of cell viability, was assessed by CellTiter-Glo assay in MCF7 and MCF7/LMTK3 cells cultured in either (A) 2D (monolayers) or (B) 3D (spheroids). All error bars represent the mean ± the standard deviation (SD) from three independent experiments. (C) Quantification in primary tumor size between MCF7 and MCF7/LMTK3 xenografts with or without doxorubicin treatment at Day 28. (Left) The means and the error bars show the standard error of the mean (SEM) for ten MCF7-LMTK3, nine MCF7-LMTK3+Dox, ten MCF7 andseven MCF7+Dox –derived xenografts. (Right) Data are displayed as individual data points together with their corresponding median values. (D) Representative images of tumors at Day 28 are shown for the different groups. Scale bars, 100 μm. (**P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001).
Fig 1.Over-expression of LMTK3 impedes the effectiveness of doxorubicin in vitro and in vivo. Following treatment with different concentrations of doxorubicin (0.05, 0.1, and 0.2 μM) for 72 h, the percentage (%) of cell viability, was assessed by CellTiter-Glo assay in MCF7 and MCF7/LMTK3 cells cultured in either (A) 2D (monolayers) or (B) 3D (spheroids). All error bars represent the mean ± the standard deviation (SD) from three independent experiments. (C) Quantification in primary tumor size between MCF7 and MCF7/LMTK3 xenografts with or without doxorubicin treatment at Day 28. (Left) The means and the error bars show the standard error of the mean (SEM) for ten MCF7-LMTK3, nine MCF7-LMTK3+Dox, ten MCF7 andseven MCF7+Dox –derived xenografts. (Right) Data are displayed as individual data points together with their corresponding median values. (D) Representative images of tumors at Day 28 are shown for the different groups. Scale bars, 100 μm. (**P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001). Furthermore, we provide details underlying the effects of LMTK3 in doxorubicin induced DNA damage/repair processes. We demonstrate that MCF7 cells stably overexpressing LMTK3 delay H2AX Ser139 phosphorylation resulting in fewer γH2AX foci as compared to parental MCF7 cells. Our analysis also reveals that ectopic expression of LMTK3 delays DNA damage response by decreasing ATM phosphorylation (Fig 2).
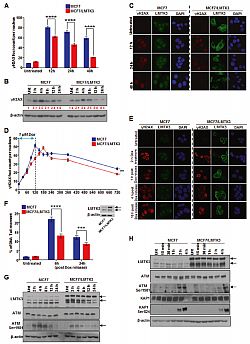
Fig. 2 Overexpression of LMTK3 delays doxorubicin-induced DNA DSBs and affects the phosphorylation of ATM. (A) MCF7 and MCF7/LMTK3 cells were treated with 0.4 μM of doxorubicin and 12, 24, and 48 h later, quantitative analysis of γH2AX foci was performed. (B) Western blot analysis for endogenous γH2AX levels in MCF7 and MCF7/LMTK3 cells following doxorubicin treatment for different time points. (C) MCF7 and MCF7/LMTK3 cells were fixed and subjected to confocal immunofluorescence analysis. Representative confocal microscopy images for γH2AX and LMTK3 are shown for different time points. (D) MCF7 and MCF7-LMTK3 cells were treated with 1 μM of doxorubicin for up to 2 h. Following, the media was removed
allowing cells to grow in fresh complete media for another 10 h. (E) MCF7 and MCF7/LMTK3 cells were fixed and subjected to confocal immunofluorescence analysis. Representative confocal microscopy images for γH2AX are shown for different time points following doxorubicin release. (F) Doxorubicin-induced DNA damage measured by neutral comet assay. MCF7 and MCF7-LMTK3 cells were treated with 1 μM of doxorubicin for up to 2 h. Following, the media was removed allowing cells to grow in fresh complete media for another 6 and 24 h. Cells were analyzed by fluorescence microscopy; (G) MCF7 and MCF7-LMTK3 cells were treated with 1 μM of doxorubicin for 2, 4, 8, 16, and 24 h. (H) MCF7 and MCF7-LMTK3 cells were treated with 1 μM of doxorubicin for 15 min, 30 min, 1, 2, and 4 h.To obtain information underlying the LMTK3-mediated doxorubicin resistance, we employed a whole genome transcriptomic analysis approach using RNA-sequencing (RNAseq). A plethora of bioinformatics analyses tools were implemented including DESeq2 (differential gene expression), the ingenuity pathway analysis (IPA; canonical pathways and disease/biological functions), the STRING database (protein–protein interaction analysis), and the Gene Ontology Enrichment Analysis (protein functions). As a result, we identified several genes and pathways that are differentially regulated by doxorubicin based on LMTK3 expression. In particular, we observed that doxorubicin treatment of LMTK3-overexpressing MCF7 cells shows a decrease in DNA damage sensing compared to parental MCF7 cells, while pathways involved in cell survival, cell viability and tumorigenesis are predominantly activated.
Finally, we examined the expression of LMTK3 in 148 pairs of primary breast cancer cases, before and after receiving chemotherapy. Interestingly, we detected a significant upregulation of LMTK3 protein levels, as assessed by immunohistochemistry, following doxorubicin and docetaxel based therapy. These results suggest that the ultimate resistance observed to these drugs could also be due to the aberrant over-expression of oncogenic LMTK3.
In summary, our data illustrate a novel role and involvement of LMTK3 in doxorubicin resistance in breast cancer. - PAPER: The structure-function relationship of oncogenic LMTK3 (Science Advances, 2020)
-
Elucidating signaling driven by lemur tyrosine kinase 3 (LMTK3) could help drug development. Here, we solve the crystal structure of LMTK3 kinase domain to 2.1Å resolution (Fig. 1), determine its consensus motif and phosphoproteome (Fig. 2), unveiling in vitro and in vivo LMTK3 substrates. Via high-throughput homogeneous time-resolved fluorescence screen coupled with biochemical, cellular, and biophysical assays, we identify a potent LMTK3 small-molecule inhibitor (C28). Functional and mechanistic studies reveal LMTK3 is a heat shock protein 90 (HSP90) client protein, requiring HSP90 for folding and stability, while C28 promotes proteasome-mediated degradation of LMTK3 (Fig. 3). Pharmacologic inhibition of LMTK3 decreases proliferation of cancer cell lines in the NCI-60 panel, with a concomitant increase in apoptosis in breast cancer cells, recapitulating effects of LMTK3 gene silencing. Furthermore, LMTK3 inhibition reduces growth of xenograft and transgenic breast cancer mouse models without displaying systemic toxicity at effective doses. Our data reinforce LMTK3 as a druggable target for cancer therapy.
 Fig 1.Crystal structure of LMTK3 Fig 2. LMTK3 phosphoproteome Fig 3. LMTK3 inhibitor model of action
Fig 1.Crystal structure of LMTK3 Fig 2. LMTK3 phosphoproteome Fig 3. LMTK3 inhibitor model of actionUltimately, the development of oral LMTK3 inhibitors may have the potential for broad clinical use, either as monotherapy or as a combinational therapy, [i.e., combined, for example, with aromatase inhibitors in ER+ BC in the same way the CDK4/6 inhibitors are and combined with the standard-of-care chemotherapy in triple-negative models]. More precisely, in the case of TNBC, despite immunotherapies being helpful at one level, there are no
approved targeted therapies. A recent report also showed promising results regarding the use of CDK12/CDK13 inhibitors alone or in combination with the chemotherapeutic agent cisplatin. Therefore, on the basis of aberrant expression of LMTK3 in TNBC and work showing that genomic inhibition of LMTK3 leads to inhibition of cell proliferation, invasion, and migration, an LMTK3 inhibitor could represent an attractive candidate for clinical trials. On the other hand, since the mechanism of endocrine and chemotherapy resistance in BC still remains largely unexplained, there remains a need to treat these patients in a more focused way, for example, in the setting of progression on CDK4/6 inhibitors. On the basis of our results so far (in vitro, in vivo, and clinical data from patient specimen cohorts), inhibition of LMTK3 may be important in tamoxifen (Tam) and doxorubicin (Dox) resensitization. Consequently, an LMTK3 inhibitor could be used alongside established therapies (e.g., Tam and Dox) to increase the sensitivity of tumors and/or potentially overcome resistance. Last, the subsequent down-regulation of ER protein levels following LMTK3 inhibition suggests additional opportunities to use LMTK3 inhibitors in patients with ESR1 mutations and/or in ESR1 fusions that may be currently untargetable.
- PAPER: LMTK3 inhibition affects microtubule stability (Molecular Cancer, 2021)
-
Pharmacological inhibition of LMTK3 (C28 treatment) leads to G2/M phase arrest of BC cells as well as MCF12A, a nontransformed breast cell line. Similarly, silencing of LMTK3 led to an increase in phospho-histone H3 (Ser10) levels, suggesting a potential role of LMTK3 in G2/M transition. Bearing in mind the effects of C28 on cell cycle arrest and the induction of cell death, we investigated whether C28 impacts microtubule dynamics, which can in turn disturb the organization of the cytoskeleton and affect cell division. Immunofluorescence of cells at the metaphase revealed that C28-treated cells present disrupted microtubule distribution and mitotic defects, including abnormal microtubule spindle organization and an altered chromosome condensation pattern.
Following, the effect of C28 on microtubule stability was confirmed using a cell-based microtubule polymerization assay, where we observed a dose dependent decrease of the insoluble polymerized tubulin fraction. Considering the role of kinase inhibitors on microtubules, we investigated the possibility that C28 is a direct tubulin-targeting agent by employing an in vitro tubulin polymerization assay. Incubation of tubulin with increasing concentrations of C28 at different time points had no effects on tubulin polymerization.
The binding affinity of C28 to tubulin was examined in vitro by monitoring the intrinsic tryptophan fluorescence of tubulin, a widely used method to determine the binding affinity of drugs for tubulin heterodimers. Our results demonstrated that although C28 can potentially bind tubulin, its relatively low binding affinity to purified tubulin dimer and the lack of any observable effect on tubulin polymerization suggest that C28 does not confer its effects by direct inhibition of tubulin polymerization. Instead our data imply that C28 modulates LMTK3-regulated pathways linked to microtubule assembly, a result that can partly explain the previously observed universal cytotoxic effects of C28 on the NCI-60 cancer cell line panel (Ditsiou et al., 2020).
In an attempt to decipher the signalling pathways via which C28 confers its effects on microtubules assembly, we used a tandem mass tagging (TMT) quantitative proteomic approach and uncovered the C28-mediated global proteomic alterations in BC cells. Interestingly, amongst the most significant C28- modulated hits was NUSAP1 (P < 0.05 and Log2 fold change of ≥|1.25|), a microtubule associated protein participating in mitotic spindle organization, whose role has been closely associated with tumour progression, chemoresistance, and poor prognosis in many tumours including BC. The effects of C28 on NUSAP1 were validated in additional BC cell lines.
Taken together, in the present study, we further demonstrated that C28 exerts its antitumor functions by degrading LMTK3, which leads to a NUSAP1-mediated microtubule instability with subsequent cell-cycle arrest and cell death. Moreover, downregulation of NUSAP1-downstream proteins involved in cell cycle regulation and microtubule stability (CDK1 and phospho-βIII tubulin), after pharmacological and genetic LMTK3 inhibition, support this hypothesis (Fig 1).
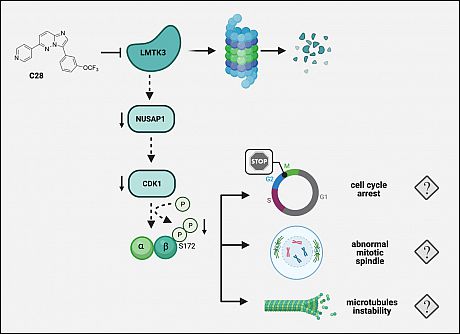
Combined, our findings propose that the pre-clinical therapeutic advantage of C28 stems from its effect on the LMTK3-targeted pathways linked to microtubule organization, acting differently from the established role of chemotherapeutic agents including vinca-alkaloids, taxanes or eribulin, which confer their cytotoxicity via their interactions with tubulin causing disruption of microtubule function. The clinical use of microtubuletargeting drugs as anticancer drugs is well-established, while the ability of standard microtubule agents (i.e. nocodazole) to modulate the activities of certain kinases has also been reported. Taken together, the development and design of multifunctional inhibitors can provide new, promising approaches for cancer treatment.
Fig 1.Schematic model depicting the proposed mechanism of action of C28 inhibitor