Glioblastoma (GBM) is the most common and fatal primary brain tumour in adults with a median survival time of 14.6 months and 5 year survival rates of less than 5% with rates decreasing with advanced age. GBM is typically classified into three subtypes, namely proneural, classical and mesenchymal, according to the different gene expression of various biomarkers including PDGFR, NF1, EGFR and CD44. Yet, despite a significant progress in describing the molecular basis of the tumour pathogenesis and consecutive standardisation of classification and treatment, GBM prognosis is still very poor.
- PAPER: Shedding of bevacizumab in tumour cells-derived extracellular vesicles as a new therapeutic escape mechanism in Glioblastoma (Molecular Cancer, 2018)
-
GBM is amongst the most aggressive types of brain tumours for which current treatments are of limited benefit. During the past decades, AAT have provided a rationale for targeting and blocking the tumour blood supply. Unfortunately, the effects of AAT/bevacizumab, a monoclonal humanised antibody neutralising Vascular Endothelial Growth Factor-A (VEGF-A), on tumour growth are short-term and GBM patients ultimately relapse. Interestingly, since the expression of some pro-angiogenic factors and their receptors (i.e. VEGF-A/VEGF-R) has been described in tumour cells, it appears that AAT/bevacizumab also acts directly on GBM cells that might eventually lead to therapy resistance and relapse.
Here, we demonstrated that Bevacizumab affects the EVs proteomic content derived from GBM cells (Fig 1).Moreover, Bevacizumab can be internalised by GBM cells (Fig 2), while it is also detectable at the surface of GBM cells-derived EVs (Fig 3).
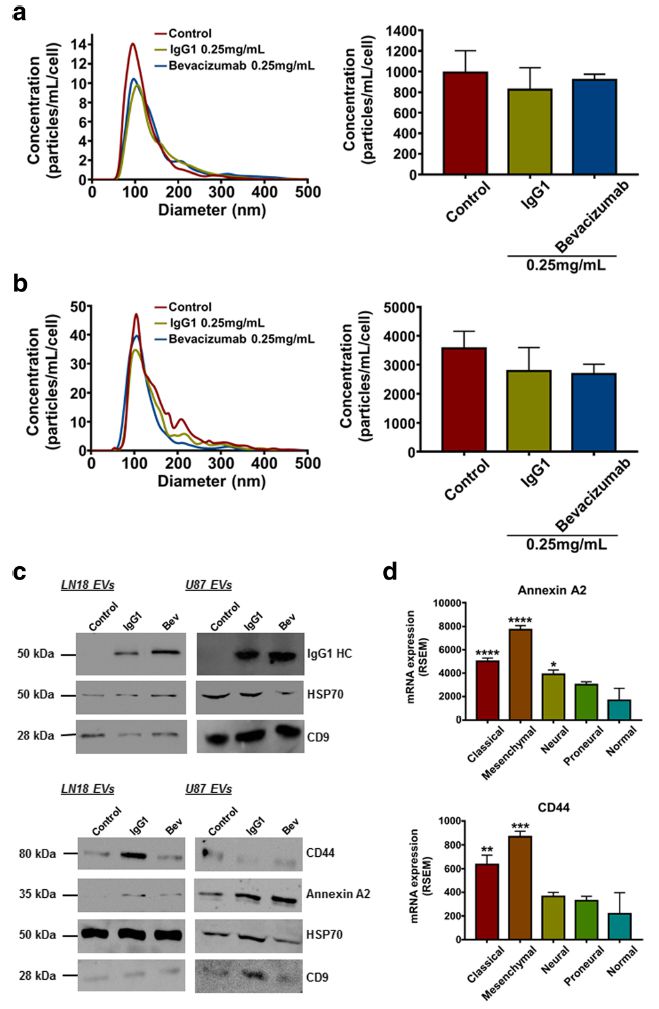
Fig. 1 IgG1/Bevacizumab antibody can affect LN18 and U87 GBM cells-derived EVs concentration and their proteomic content. NTA of (a) LN18 or (b) U87 GBM cells-derived EVs following treatment with bevacizumab (0.25 mg/mL). LN18 or U87 GBM cells were treated for 24 h with 0.25 mg/ mL IgG1 or bevacizumab. Then cells were washed two times with sterile PBS and incubated additional 24 h in serum free conditions without treatment. CM was then collected, EVs were isolated and re-suspended in 100 μL filtered sterile PBS. EVs suspension was 1/5 diluted and infused to a Nanosight© NS300 instrument. 5 captures of 60s each were recorded. Particles concentration (particles/mL) and size (nm) were measured. Particles concentration was normalised to the number of cells after treatment (particles/mL/cell). The mean ± SEM of 3 independent experiments is shown. (c) Western blotting validation of the human IgG, Annexin A2 and CD44 in EVs derived from LN18 and U87 GBM cells. (d) Gene expression distribution of Annexin A2 and CD44 among the different GBM subtypes has been obtained from TCGA. The mean ± SEM is shown (*p < 0.5, **p < 0.01, ***p < 0.001,****p < 0.0001; ANOVA, compare to ‘normal’).
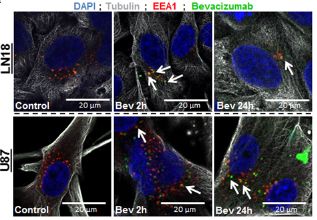
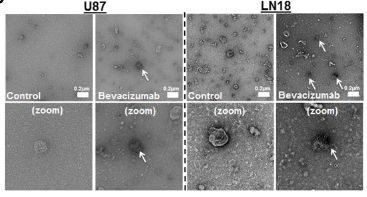
Fig. 2 (upper left)Bevacizumab is internalised by GBM cells Immunofluorescence detection of bevacizumab and EEA1 in LN18 and U87 GBM cells. GBM cells were allowed to grow on cover slips and then treated with 0.25 mg/mL bevacizumab for 2h and 24h. Cells were fixed with 4% PFA and then incubated with antibodies against α-tubulin, EEA1 and human IgG1.
Fig. 3 (upper right) Bevacizumab is detectable at the surface of GBM cells-derived EVs following treatment TEM detection of bevacizumab in LN18 and U87 GBM cells-derived EVs. U87 GBM cells were treated for 24 h with 0.25 mg/mL bevacizumab. Then cells were washed two times with sterile PBS and incubated additional 24 h in serum free conditions without treatment. CM was then collected and EVs were isolated. Immuno-gold labeling was then performed against human IgG in the EVs fractions.
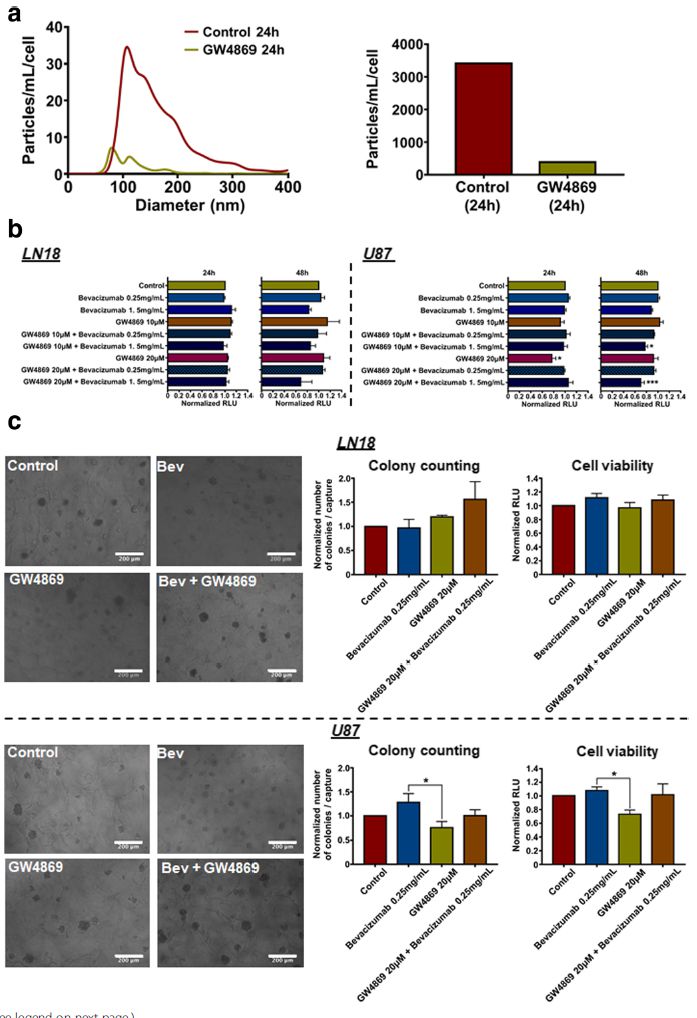 Moreover, we provide evidence showing that inhibition of EVs production increases the effects of bevacizumab on GBM cells’ viability (Fig 4). Taken together, we report that the paradoxical pro-invasive effect of bevacizumab on GBM cells might be due to alterations in the tumour cells-derived EVs, including shedding of the antibody and further modifications of the cargo, both possibly contributing to therapeutic resistance. Therefore, this study suggests the combination of bevacizumab with a local blocking of EVs-dependent intercellular communication as a potential new therapeutic strategy to improve GBM treatment.
Moreover, we provide evidence showing that inhibition of EVs production increases the effects of bevacizumab on GBM cells’ viability (Fig 4). Taken together, we report that the paradoxical pro-invasive effect of bevacizumab on GBM cells might be due to alterations in the tumour cells-derived EVs, including shedding of the antibody and further modifications of the cargo, both possibly contributing to therapeutic resistance. Therefore, this study suggests the combination of bevacizumab with a local blocking of EVs-dependent intercellular communication as a potential new therapeutic strategy to improve GBM treatment.Fig. 4 Inhibition of EVs production increases effects of bevacizumab on U87 GBM cells' viability. (a) NTA of LN18 GBM cells-derived EVs following treatment with GW4869 (20 μM). Particles concentration was normalised to the number of cells after treatment (particles/mL/cell). (b) LN18 and U87 GBM cells' viability assay in response to bevacizumab combined with GW4869. Results are expressed as normalised Relative Light Unit (RLU). (*p < 0.05, ***p < 0.01; ANOVA). (c) LN18 and U87 GBM cells' invasiveness assay using a hyaluronic acid (HA) hydrogel. Results are expressed as normalised number of colonies / capture and normalised RLU, respectively. (*p < 0.05; ANOVA).
- PAPER: Definition of an inflammatory biomarker signature in plasma-derived extracellular vesicles of Glioblastoma patients (Biomedicines, 2022)
-
The complex interactions between cancer cells and their surrounding microenvironment are amongst the parameters that make GBM treatment challenging. Extracellular vesicles (EVs) represent one of the means of such tight intercellular communication. We and others have observed that the cargo of sEVs can mirror the molecular background of their GBM cell of origin, consequently providing crucial information regarding the associated molecular subtype, the tumour aggressiveness and the response to treatments. For these reasons, EVs have gained increased interest over the last decade as potential biomarker candidates for diagnosis, disease recurrence and monitoring response to treatment in GBM patients. Currently, serial brain imaging is used for detecting tumour progression. However, this modality is challenging, as it is sometimes difficult to determine true progression from pseudo-progression. On the other hand, circulating EVs have very short half-life in vivo and, therefore, can be used in identifying rapid changes in tumour progression. This method could prove invaluable and perhaps identify tumour progression at a stage which is undetectable by conventional imaging.
In this study, we isolated sEVs from blood liquid biopsies of GBM patients and characterised their proteomic cargo to identify potential candidate GB biomarkers. An inflammatory biomarker signature was described in sEVs from GBM patients. Such application would be invaluable in future clinical practice for diagnosis and fine-tuning of GBM treatment.
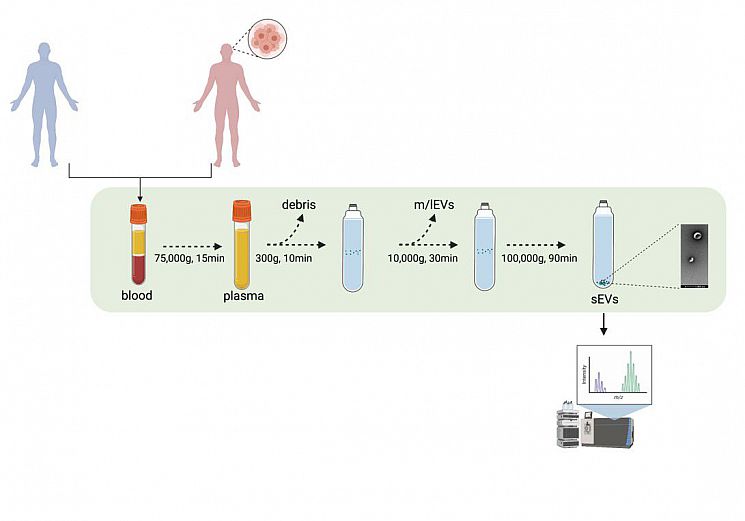 Fig. 1 Experimental pipeline. sEVs from GB patients and healthy volunteers’ blood samples were isolated through UC. The proteomic content of the sEVs were deciphered using mass spectrometry.
Fig. 1 Experimental pipeline. sEVs from GB patients and healthy volunteers’ blood samples were isolated through UC. The proteomic content of the sEVs were deciphered using mass spectrometry.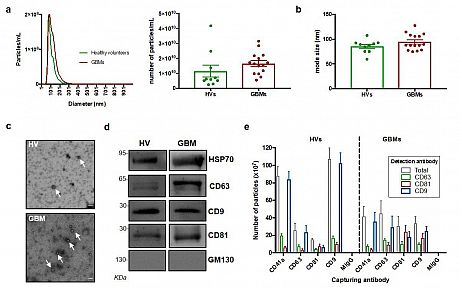
Fig. 2 Characterisation of sEVs isolated from GBM patients and healthy volunteers. (a) NTA of sEVs. The sEV suspensions from healthy volunteers (HV) (n = 10) and GBM patients (n = 15) were diluted 1:50 and infused into a NanoSight NS300 instrument. Five captures of 60 s each were recorded. Particle concentration (particles/mL) and size (nm) were measured. The mean number of particles/mL + SEM is shown. (b) Mode size (nm) ± SEM of sEVs is shown (healthy volunteers = 10; GB patients = 15). (c) Representative electron microscopy images of GB patient- and healthy volunteer- derived sEVs; 30k magnification was used. White arrows show sEVs. Scale bar = 100 nm. (d) Representative Western blots of GBM patient- and healthy volunteer-derived sEVs, showing the presence of protein markers commonly associated with sEV subpopulations. The absence of the Golgi protein marker, GM130, indicates that non-EV cellular components were below the detection threshold. The same protein amounts of sEVs (10 μg) were loaded per lane. (e). ExoView analysis of sEV markers. sEV samples were incubated on microarray chips coated with the indicated antibodies. CD9-, CD63- and CD81-positive particles were detected after probing with a cocktail of fluorescent tetraspanin antibodies using the ExoView R100 platform. Results are the average of 2 healthy volunteer- and 2 GB patient-derived samples. For each sample, three technical replicates were performed. All data were adjusted for the dilution of the sample onto the chip. **** p < 0.0001.
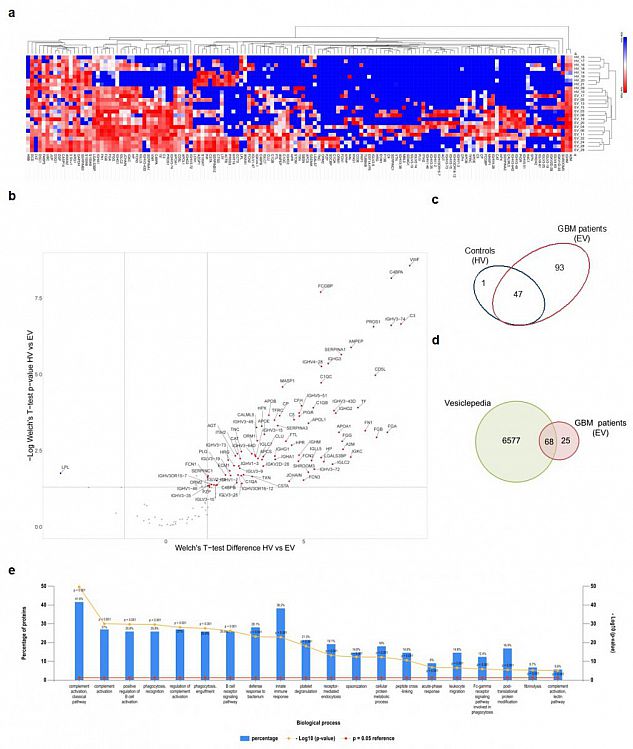 Fig. 3 Proteomic analysis of sEV cargo. (a) Hierarchical clustering of protein expression in all experimental samples (i.e., healthy volunteers (HVs) and GBM patients (EVs)) using Euclidian distance and average linkage, showing a heat map of protein expression for the respective samples (no protein expression in blue to high protein expression in red) and dendrograms for the hierarchical clustering of samples (horizontal dendrogram) and for proteins (vertical dendrogram), respectively. (b) Volcano plot showing the differentially expressed proteins between HVs and EVs with Welch’s t-test p-values < 0.05 and a log2 Welch’s t-test difference of at least 1.5 for upregulated proteins (red dots) and −1.5 for downregulated proteins (blue dots). (c) Venn diagram showing the repartition of the identified MS hits between HVs and EVs. (d) Venn diagram of proteins enriched in GBM-derived sEVs compared with proteins annotated in the Vesiclepedia database. (e) Gene enrichment analysis for “biological process” was performed based on MS hits using the FunRich platform. The 20 most significant processes identified are reported.
Fig. 3 Proteomic analysis of sEV cargo. (a) Hierarchical clustering of protein expression in all experimental samples (i.e., healthy volunteers (HVs) and GBM patients (EVs)) using Euclidian distance and average linkage, showing a heat map of protein expression for the respective samples (no protein expression in blue to high protein expression in red) and dendrograms for the hierarchical clustering of samples (horizontal dendrogram) and for proteins (vertical dendrogram), respectively. (b) Volcano plot showing the differentially expressed proteins between HVs and EVs with Welch’s t-test p-values < 0.05 and a log2 Welch’s t-test difference of at least 1.5 for upregulated proteins (red dots) and −1.5 for downregulated proteins (blue dots). (c) Venn diagram showing the repartition of the identified MS hits between HVs and EVs. (d) Venn diagram of proteins enriched in GBM-derived sEVs compared with proteins annotated in the Vesiclepedia database. (e) Gene enrichment analysis for “biological process” was performed based on MS hits using the FunRich platform. The 20 most significant processes identified are reported.
- PAPER: PDGF-R inhibition induces glioblastoma cell differentiation via DUSP1/p38MAPK signalling (Oncogene, 2022)Oncogene, 2022)
-
While numerous therapeutic approaches aim for cancer cell death, a few studies have instead started exploring treatments that induce cancer cell differentiation into non-neoplastic cells as a novel strategy. GBM treatment could highly benefit from differentiation therapies. High resistance to classic cytotoxic treatments and the presence of pluripotent cancer stem cells (CSCs) in residual tumour tissues due to incomplete surgical excision make GBM a good candidate for differentiation therapies. Several potential molecular targets of interest have thus been tested and reported for cancer cell differentiation therapy in GBM, including activators of cyclic adenosine monophosphate (cAMP), which reverse the Warburg effect and cause a metabolic shift that drives differentiation of tumour cells into astroglia.
In addition, as kinases are involved in the development and progression of cancer, including GBM, and due to their activity in regulating differentiation, recent reports have suggested targeting kinases in differentiation therapies against GBM. Kinase inhibitors (KIs) with the potential to induce differentiation in GBM include aurora-A inhibitors alisertib and MLN8237, which were able to induce senescence, differentiation, and cytotoxicity in a GBM neurosphere model and GBM cells in vitro, respectively. In accordance with these reports, the present study describes the screening of a panel of KIs that led to the identification of a receptor tyrosine kinase (RTK) inhibitor, namely CP-673451, which targets the platelet-derived growth factor receptor α/β (PDGF-Rα/β) as a potential differentiation agent in GBM. Herein, we propose that CP-673451 treatment can induce the upregulation of phosphatase dual-specificity phosphatase 1 (DUSP1) and consecutive downregulation of phosphorylated p38 mitogen activated protein kinase (MAPK), leading to the onset of differentiation of GBM cells, thus limiting proliferation and invasion in vitro, while improving the anti-tumour effects of temozolomide in vivo.
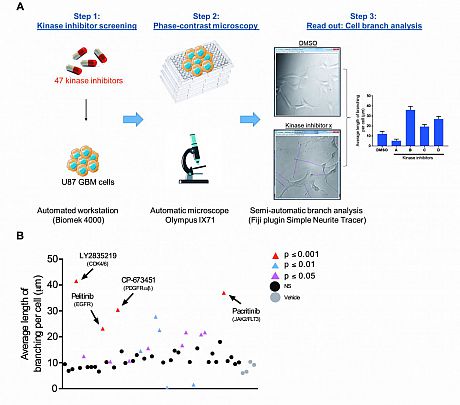 Fig. 1 Kinase inhibitor screening identifies agents that induce neurite-like process outgrowth in U87 GBM cells. (A) Step 1: U87 GBM cells were seeded into 96-well plates using the Biomek 4000 automatic workstation. Following incubation for 24 h, cells were treated with 1 μM of the selected KIs (n = 47). Step 2: After 24 h phase-contrast microscopy images were taken at 20x mag using the Olympus IX71 microscope (Micromanager software). Step 3: The length of long thin extensions termed ‘neurite-like process’ were analysed, using the Fiji plugin ‘simple neurite tracer’ (Image J) and the average length neurite-like process per cell determined. (B) U87 GBM cells were treated for 24 h with 1 μM 47 kinase inhibitors. The average length of neurite-like process was measured and normalised by cell number. The mean ± SD of n = 3 independent experiments is shown. Grey circles vehicle control, black circles not significant (NS) p ≥ 0.05, pink triangles p ≤ 0.05, blue triangles p ≤ 0.01, red triangles p ≤ 0.001 (two-tailed t-test). Kinase inhibitor ‘hits’ from the screening p ≤ 0.001 are indicated with black arrows and include; LY2835219 (CDK4/6), pelitinib (EGFR), CP-673451 (PDGF-Rα/β) and pacritinib (JAK2/FLT3).
Fig. 1 Kinase inhibitor screening identifies agents that induce neurite-like process outgrowth in U87 GBM cells. (A) Step 1: U87 GBM cells were seeded into 96-well plates using the Biomek 4000 automatic workstation. Following incubation for 24 h, cells were treated with 1 μM of the selected KIs (n = 47). Step 2: After 24 h phase-contrast microscopy images were taken at 20x mag using the Olympus IX71 microscope (Micromanager software). Step 3: The length of long thin extensions termed ‘neurite-like process’ were analysed, using the Fiji plugin ‘simple neurite tracer’ (Image J) and the average length neurite-like process per cell determined. (B) U87 GBM cells were treated for 24 h with 1 μM 47 kinase inhibitors. The average length of neurite-like process was measured and normalised by cell number. The mean ± SD of n = 3 independent experiments is shown. Grey circles vehicle control, black circles not significant (NS) p ≥ 0.05, pink triangles p ≤ 0.05, blue triangles p ≤ 0.01, red triangles p ≤ 0.001 (two-tailed t-test). Kinase inhibitor ‘hits’ from the screening p ≤ 0.001 are indicated with black arrows and include; LY2835219 (CDK4/6), pelitinib (EGFR), CP-673451 (PDGF-Rα/β) and pacritinib (JAK2/FLT3).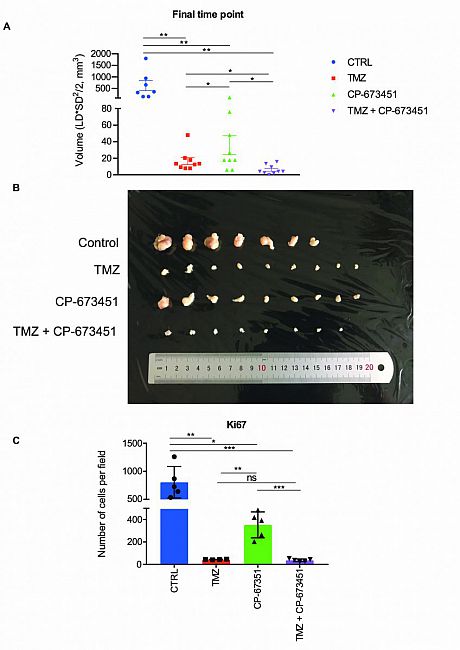
Fig. 2 Combination with CP-673451 improved anti-tumour effect of temozolomide in a subcutaneous GBM mouse model.
(A) Volumes of U87 GBM cell subcutaneous xenograft tumours from athymic nude mice were measured after 3 weeks of treatment with TMZ (25 mg/kg), CP-673451 (40 mg/kg) or combination treatment. The mean ± SEM is shown. (B) Image of the tumours extracted from mice after 3 weeks of treatment separated into each treatment group. C Ki67 immunohistochemistry staining was performed on tumour tissue post treatment and the number of cells stained per field quantified. The mean ± SD is shown *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001 (two-tailed t-test).
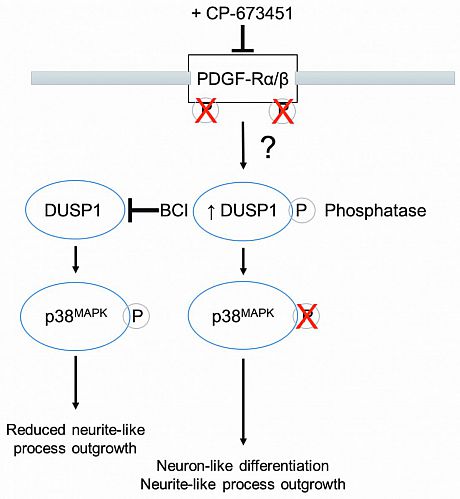
Fig. 3 PDGF-Rα/β inhibition by CP-673451 induces differentiation of glioblastoma cells via DUSP1/p38MAPK pathway activation. Diagram of potential mechanism of action of CP-673451 in GBM cells.