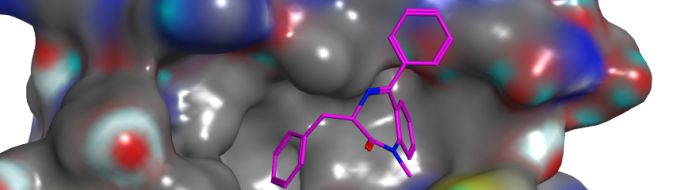
To support activities in the Translational Drug Discovery Group at Sussex we have established computational biology and chemistry protocols to support the effective design of novel chemical modulators. Cheminformatic protocols have been designed for activities such as compound library selection, virtual screening, scaffold hopping and rational drug design. We also have an established expertise in protein modelling and small molecule docking.
Computational approaches fully exploit the structural data provided by the structural biology teams within the Genome Damage and Stability Centre at Sussex, who utilise X-ray crystallography to characterise protein-ligand interactions. In addition to these activities, databases are being developed to integrate biological and chemical data to support the drug discovery process.