Research
DNA repair mechanisms & chromosome stability group
We focus on different aspects of genome stability, in particular: (1) mechanisms of chromosome break repair, (2) maintenance of chromosome stability during genome replication, and (3) links to human disease and biomedical implications.
Recent research highlights, explained in greater detail below, include novel insights into the early events of DNA double-strand break repair and the identification of new DNA replication stress response pathways, which promote full genome replication and the resolution of post-replicative sister chromatid links in time for chromosome segregation.
Mechanisms of chromosome break repair
DNA double-strand breaks disrupt the continuity of chromosomes and can give rise to gross chromosomal aberrations, such as those frequently seen in cancer cells. In contrast to the DNA ends found at the termini of chromosomes, which are protected by proteinaceous structures known as telomeres, DNA double-strand break ends provoke a DNA damage response and are channeled towards repair by a splicing mechanism (non-homologous end-joining; NHEJ) or homology-directed repair (homologous recombination; HR). Intriguingly, the protein Rif1 (Rap1-interacting factor 1) has been implicated at telomeres as well as DNA breaks. First, Rif1 was identified as a telomeric protein in S. cerevisiae, and later as a key mediator of DNA double-strand break repair pathway choice (NHEJ vs. HR) in mammals, a functional dichotomy that has long been a puzzle for the field. In collaboration with the groups of Nicolas Thomä (FMI, Basel) and David Shore (University of Geneva), we have elucidated Velcro-like protein-protein interactions between Rif1, Rap1, and Rif2 at yeast telomeres (Shi et al., Cell 2013). Subsequently, we uncovered a novel DNA-binding site within a hook-shaped Rif1 domain, which has the ability to fully encapsulate DNA ends, forming a protective protein sheath that counteracts telomerase and checkpoint signaling at uncapped telomeres. At chromosome breaks, we revealed that direct DNA binding by Rif1 attenuates DNA end-resection, stabilizing DNA ends for repair by NHEJ. These findings provide a pleasing unification of the yeast and human systems and identify exclusion of end-processing factors as an underlying mechanistic principle for diverse biological roles of Rif1 in telomere homeostasis and DNA repair (Mattarocci et al., Nature Structural & Molecular Biology 2017; Fontana et al., Microbial Cell 2018). Current projects focus on the spatiotemporal regulation of chromosome break repair.
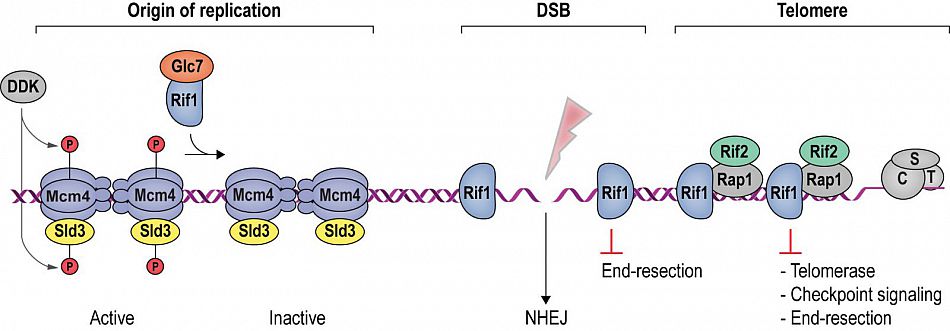
Rif1 is a multi-faceted genome maintenance factor. At origins of replication, Rif1 attenuates origin firing by recruiting protein phosphatase 1 (Glc7), which reverses activating phosphorylation events on components of the replication machinery. At DNA double-strand breaks (DSBs), Rif1 tightly encases the break ends, gating access of end-resection factors. As a result, DSB ends are stabilized, promoting their re-ligation by NHEJ. At budding yeast telomeres, Rif1 forms part of an interlinked telosome protein meshwork with Rap1 and Rif2, underpinning telomere architecture and function. Fontana et al. (2018), Microbial Cell, 5 (7). pp. 327-343.
Maintaining chromosome stability during DNA replication and chromosome segregation
In eukaryotes, DNA replication initiates at multiple origins of replication distributed along the chromosomes. Activated origins give rise to bidirectional replication forks, driven by multi-subunit protein machines (known as replisomes) that mediate DNA synthesis. DNA damage, transcription, and a number of additional factors can perturb replication fork progression (causing replication stress), and arrested replication forks may lose their activity and collapse. This can lead to genome underreplication, jeopardizing chromosome segregation and chromosome stability. Two principle activities, initiated by the replication checkpoint, ameliorate replication stress. First, activation of dormant origins near arrested forks and a delay in origin firing elsewhere reduces global replication fork stalling while compensating for any replication shortfall locally. Secondly, DNA repair pathways become activated that mediate protection and restart of stalled replication forks. The molecular mechanisms underlying these fundamental processes at DNA replication forks remain incompletely understood. We have recently identified a two-tiered replication stress response involving disease-linked nuclease-helicase Dna2 and structure-specific nuclease/Holliday junction resolvase Yen1 (GEN1 in human, see Ip et al., Nature 2008). Jointly, Dna2 and Yen1 promote full genome replication and the resolution of dead-end replication intermediates in mitosis to ensure cell viability (Ölmezer et al., Nature Communications 2016; Falquet and Rass, Microbial Cell 2017). Our studies help unravel the pathologies associated with Dna2, which is implicated in cancer, mitochondrial myopathy, and the primordial dwarfism disorder Seckel syndrome. Moreover, our findings provide new entry points to explore at the molecular level how cells achieve complete chromosome replication in every cell cycle.
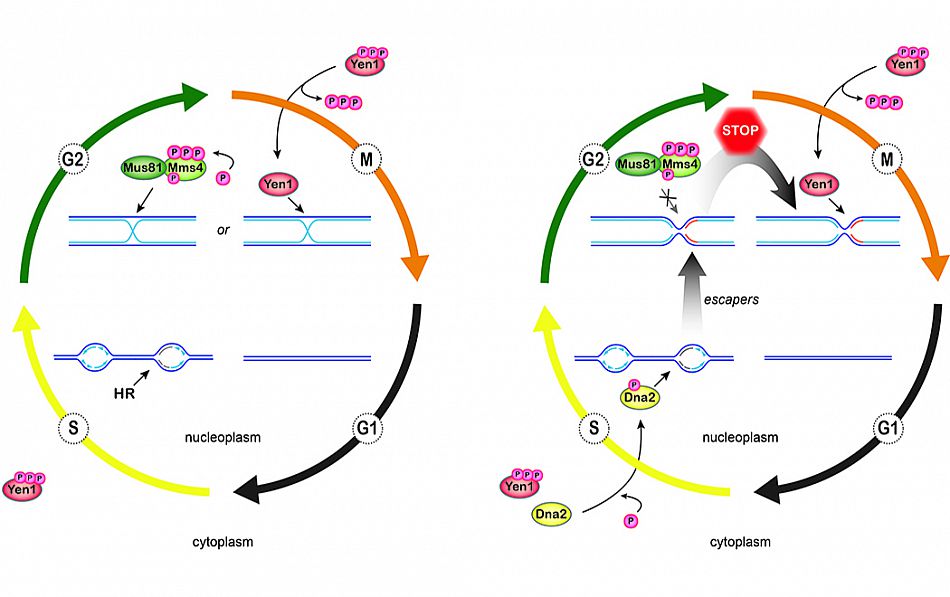
Model for a two-tiered response to replication stress by Dna2 and Yen1. The canonical role of Yen1 in the resolution of homologous recombintion (HR) intermediates is shown on the left. Mus81-Mms4 is activated by hyperphosphorylation in G2/M, prior to activation and nuclear import of Yen1 upon anaphase onset. Thus, HR intermediates are predominantly cleaved by Mus81-Mms4, with Yen1 acting as a catchall in M phase to remove persistent recombination structures in time for chromosome segregation. Right: Parallel to HR, the Dna2 helicase is tending to stalled replication forks. Replication intermediates that escape the attention of Dna2 give rise to toxic structures that are sensed by the DNA damage checkpoint, but which are refractory to processing by Mus81-Mms4. At anaphase entry, Yen1 is activated and uniquely resolves persistent replication intermediates, averting mitotic catastrophe. Ölmezer et al. (2016), Nature Communications 7. Article number: 13157.
Biomedical implications: Replication stress and cancer
Precision medicines play an increasingly important role in cancer therapy. They offer molecularly targeted approaches that are less pleiotropic than conventional chemo- and radiotherapies, which act indiscriminately on normal and cancerous cells. The design of targeted cancer therapies requires mechanistic insight into cancer-specific processes. While replication stress is not unique to cancer cells, hyperproliferating cancer cells have higher stress levels than normal cells. Targeting mechanisms that mediate replication fork recovery might thus provide opportunities to selectively eliminate cancer cells by stress-overload. In collaboration with the group of Momo Bentires-Alj (University Hospital Basel), we have developed a pathophysiologically relevant breast cancer model as a platform that allows us to assess the effects of targeted perturbations of the replication stress response network on cancer cell growth and tumour formation. We pursue targets identified in the literature and by our own mechanistic studies. Since the concept of exploiting replication stress for therapy is not specifically tied to one particular cancer type, broad relevance of our findings can be expected.